Yves here. KLG here describes another success of biomedical science against a genetic disease, here cystic fibrosis, and why that success has been less complete and harder to achieve than against sickle cell anemia and hemophilia but also describes the price gouging that has also resulted.
By KLG, who has held research and academic positions in three US medical schools since 1995 and is currently Professor of Biochemistry and Associate Dean. He has performed and directed research on protein structure, function, and evolution; cell adhesion and motility; the mechanism of viral fusion proteins; and assembly of the vertebrate heart. He has served on national review panels of both public and private funding agencies, and his research and that of his students has been funded by the American Heart Association, American Cancer Society, and National Institutes of Health.
Last time here we discussed recent developments in the treatment of two genetic diseases: hemophilia and sickle cell disease. Gene therapy for Hemophilia A and B, which are clinically indistinguishable blood clotting disorders caused by mutations in two different genes, is now likely to be a specific, long-lasting treatment of choice for each disease. This comes about 40 years after the two genes responsible for HA and HB were identified. The mutant gene responsible for sickle cell anemia was identified in 1949. In 2019, 70 years after the discovery that sickle cell disease is caused by a single mutation in the beta chain of the oxygen-carrying, red protein hemoglobin, a specific drug treatment was identified. The overt pathology of sickle cell disease, the deformation of erythrocytes into sickle-shaped red blood cells, is caused by the polymerization of mutant hemoglobin into long fibers. Treatment with voxelotor, which is tolerated at high doses in children and adults in the form of a pill taken orally prevents hemoglobin polymerization and thus the disease.
Both of these treatments are triumphs of biomedical science. Although slow in development they represent specific treatments of devastating genetic diseases [1], which brings us to anther disease caused by mutations in a single gene, cystic fibrosis. CF is second to sickle cell disease on the list of common genetic diseases that are homozygous recessive (two mutant copies of the gene are required for the disease to appear).
Cystic fibrosis is caused by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) in those who inherit two mutant copies of the gene for CFTR. The most common CFTR mutation, which is responsible for 70% of CF cases, is on one level seemingly inconsequential. Normal CFTR has 1,480 amino acids. The CF mutant CFTR has 1,479 amino acids, lacking the phenylalanine (F) at position 508 (sequence alignment below, deletion circled [2]). Thus, the mutant protein is 99.93% identical to the normal protein. Fine details matter at the molecular level, as they do everywhere else. The gene for CFTR was first cloned in 1989, an event accompanied by great excitement. CF was one of the earliest diseases for which the gene was cloned, and it was expected that a specific therapy for CF would follow in due course, perhaps within a few years. This did not happen.

The most widely appreciated symptom of CF is mucous congestion in the lung that leads to persistent infections. An affecting account of CF is found in Alex: the Life of a Child (orig. 1983), the eminent sportswriter Frank Deford’s book about his daughter Alex who died of CF at eight years old in 1980. As clinical interventions and symptom management have improved, people with CF have lived much longer on average. In the 1960s life expectancy of CF patients was less than 20 years, and I knew the slightly older brother of a friend whose CF left him slight in stature. He was the accomplished trumpet player in the marching band but died shortly after graduating from high school. CF life expectancy was 38 years ten years ago and stands at 53 years today for those born between 2017 and 2021. Progress, indeed.
Why has gene therapy for CF not come to fruition? The primary reasons are found in the previous description of gene therapy for hemophilia. The coagulation factors F8 (HA) and F9 (HB) are produced in the liver and secreted into the circulation where they participate in blood clotting. Thus, the target for this gene therapy is the liver. Stable transduction of liver cells by Adeno-Associated Virus-5 (AAV-5) carrying the gene for F8 or F9 is simple and straightforward. Active F8 or F9 protein is secreted into the blood by these transduced liver cells for at least five years and the underlying hemophilia is cured during this time.
A similar strategy for the lung is much more problematic for a host of technical reasons, but CFTR is required in organs other than the lung even though lung congestion due to excessive mucous secretion and bacterial infections are the most visible targets for CF symptom management. CFTR deficiency also causes problems in the pancreas. Early in life exocrine pancreatic failure results in intestinal maldigestion and malabsorption followed by growth failure because digestive enzymes are not properly delivered to the small intestine. Later in life CF-related diabetes is caused by endocrine failure of pancreas in which the antagonist hormones insulin and glucagon are not produced, leading to disrupted metabolism. CF is also associated with “bowel problems, liver disease, reproductive disfunction, sinus disease, bone disease and elevated sweat chloride concentrations.” Babies with CF are often diagnosed because they “taste” salty when kissed by their parents; salt is sodium chloride. So, gene therapy for CF would have to target virtually the entire body to work well.
Nevertheless, the cloning of CFTR and 30 years of research on its function have led to clinical interventions that are very promising. But first, understanding this requires a very short lesson in cell biology. CFTR is found in the cell surface membrane (plasma membrane), where it functions as a chloride channel (cells “want” the chloride concentration to be higher on the outside than inside). The major CFTR mutant (Phe508del) does not fold properly on its way to the membrane and gets stuck in the pathway and degraded. The small amount of mutant CFTR that does get to the membrane displays defective gating (all channels for all things that pass through the membrane are gated, which is another way of saying “regulated”). So, a compound that helps mutant CFTR fold properly and get to the cell membrane would lessen the deleterious effects of the mutant, provided that the protein could function when it got there. Figure 1 of the paper linked above illustrates this. The common CFTR mutant is a Class II mutant (Phe508del). If this compound, or another, also improved the gating of the CFTR, CF pathology would be further lessened.
Based on this reasoning two classes CFTR modulators have been developed. The first are called “correctors,” or chemical chaperones [3]. These compounds help a protein find its proper conformation (three-dimensional shape) so that it can be processed and trafficked to the cell surface. The other modulators are called “potentiators.” These improve the chloride channel function of mutant CFTR when it gets to the cell membrane. A corrector and a potentiator acting together will improve the function of CFTR(Phe508del) and other mutants.
Vertex Pharmaceuticals screened thousands of compounds to identify CFTR correctors and potentiators. The first was the potentiator ivacaftor (Kalydeco) that increased the time the CFTR channel remains open at the cell surface. This resulted in significant improvement in lung function in CF patients with the Class III mutation Gly551Asp (glycine at position 551 changed to aspartic acid; this is a much rarer CF mutant than Phe508del). Patients treated with ivacaftor were less likely to be admitted to the hospital and had lower rates of Pseudomonas (bacterial) and Aspergillus (fungal) infections compared to controls. They also maintained lung function better and for longer periods than untreated patients.
The first combination therapy of a corrector and potentiator (ivacaftor plus lumacaftor; Orkambi) showed measurable but marginal improvements in lung function in Phe508del CF patients, with liver dysfunction as a major side effect. But as with virtually all biomedical science, the development of these drugs has led to incremental and significant improvements in lung function in CF patients. The current triple therapy [4] consisting of two correctors elexacaftorand tezacaftor, plus the original potentiator ivacaftor (Kaftrio in the UK and Trikafta in the US) substantially increases the amount of mutant CFTR at the cell surface and is indicated for treatment of CF in Classes II-V, representing about 90% of CF patients.
Based on current results, this triple therapy of elex/tez/iva (lightly edited, emphasis added):
has the ability to transform outcomes for the vast majority of people with CF. The magnitude of improvement in FEV-1 (lung function), the decrease in pulmonary exacerbations (mucous buildup and bacterial/fungal infections), the nutritional improvement, and improved quality of life are unparalleled. Most people with CF receiving (these) modulators are likely to have a sweat chloride concentration below the diagnostic threshold for CF. Although superseded by elex/tez/iva (Kaftrio/Trikafta), iva alone (Kalydeco), lum/iva (Orkambi), and tez/iva may still have a role in certain variants of CF (Table 1)…CFTR modulators are likely to be the most important development in CF care for a generation, and possibly forever. For a disease that used to be universally fatal in childhood, they offer hope to a majority of people with CF and they families, and their advent should be seen as a new dawn.
These are unusually strong statements for a scientific review. But I have been following this clinical literature since the first CFTR modulators were introduced (and initially denied approval in the UK because of its uncertain effectiveness and cost) and the enthusiasm is warranted in my view. I also did what should be standard for anyone reading the biomedical literature by checking the acknowledgments and disclosures. There was nothing much to see; internal peer review by the editors of BMJ seems appropriate for this paper:
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors
Competing interests: None declared
Provenance and peer review: Commissioned; internally peer reviewed.
The benefits of modulator therapy for CF are now clear. These drugs in combination can be used to treat multiple classes of CF. Moreover, while CFTR-Phe508del is most common in populations with origins in Western and Northern Europe, other forms of CF more prevalent in the rest of the world can also be treated similarly. These rare variants may not be orphan diseases after all. So, what is not to like? Cost. As mentioned above, the first modulator treatments for CF were not approved in the UK because it was not clear that the benefits outweighed the costs.
The price of Tricafta in the US was $311,000 per patient per year in 2021 and seems to have stayed in that range, but determining the actual cost of the drug is difficult if not impossible. There are about 40,000 CF patients in the United States with about 105,000 diagnosed in 94 countries according to the Cystic Fibrosis Foundation. In 2021 that $311,000 comes out to $852 per day per patient. Or $34.1 million per day nationwide, about $12.4 billion per year in potential sales.
There is no question that Vertex Pharmaceuticals has done great work with these drugs. But naturally their prices have been addressed in multiple forums. Here, for example. A group of scientists from Imperial College London, University of North Dakota, Howard University, and University of Liverpool has also looked systematically at the cost of production of the CFTR modulators marketed by Vertex. Directly from their structured Abstract (emphasis added):
Methods: Minimum costs of production for CFTR modulators were estimated via an algorithm validated in previous literature and identification of cost-limiting key starting materials from published routes of chemical synthesis. This algorithm utilised per kilogram active pharmaceutical ingredient costs obtained from global import/export data. Estimated production costs were compared with published list prices in a range of countries.
Results: Costs of production for elexacaftor/tezacaftor/ivacaftor are estimated at $5,676 [$4,628-6,723] per year, over 90% lower than the US list price. Analysis of chemical structure and published synthetic pathways for elexacaftor/tezacaftor/ivacaftor revealed relatively straightforward routes of synthesis related to currently available products. Total cost of triple therapy for all eligible diagnosed CF patients worldwide would be $489 million per year. Comparatively, the annual cost at US list price would be $31.2 billion.
A picture is worth several thousand words and even more money. Their Figure 2(C) below shows the prices of a yearlong course of elex/tez/iva (Kaftrio/Trikafta) as of December 2021 in the US, selected countries in Europe, and Argentina and compares these prices to the cost of generic production of the three modulators (Estimated Cost, far right).
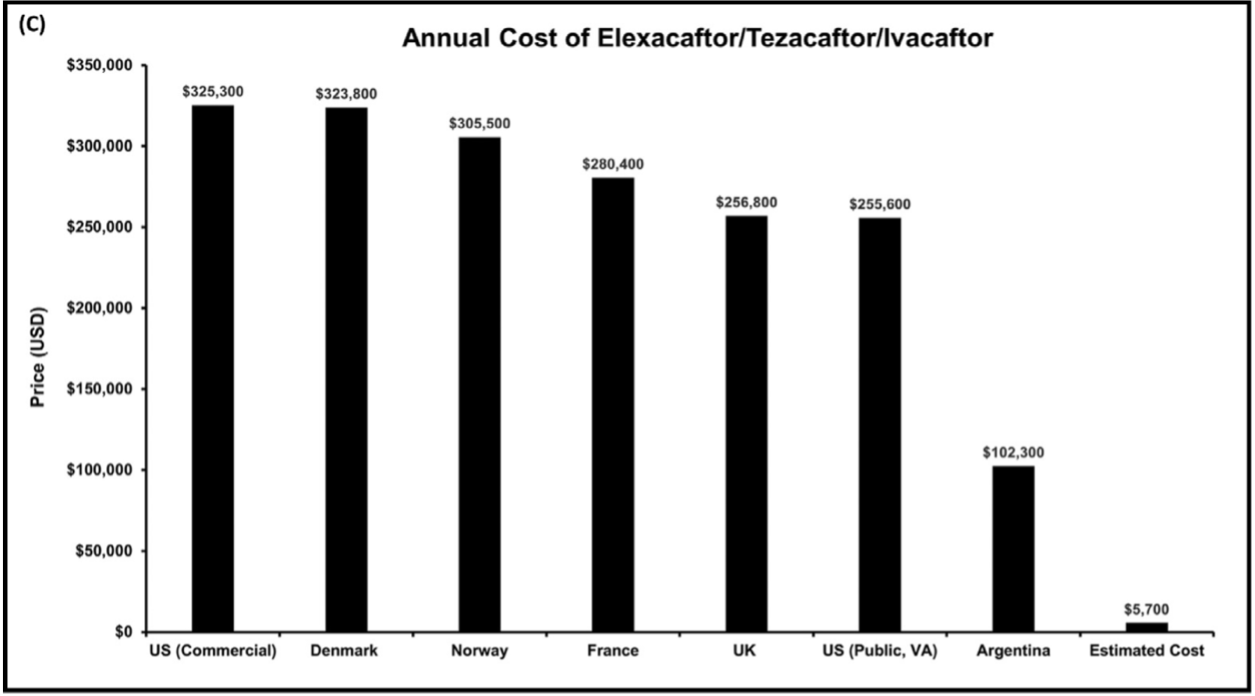
In the US, that is $325,300/$5,700. Thus, the markup for the US commercial price is, according to my ancient and irreplaceable HP33s calculator, approximately 5,607%, or a factor of 57. And this is not an uncommon thing, as has been shown by recent critical studies of Biomedicine and its adjunct Big Pharma, here and here and here (the latter reviewed here). One cannot help but think of Jonas Salk, who when asked if the polio vaccine had been patented, replied “Could you own the sun?” And Banting, Best, and Macleod, who “gave” insulin to the University of Toronto for one dollar so that it would be available to those in need in perpetuity.
And therein lies the difference between Biomedical Science and Biomedicine. The one has great power and utility, when practiced properly and developed as a public good. The other also has great power but the results are distributed much more unevenly than necessary while perched on an essential foundation in biomedical science that was funded primarily by the public as a public good.
A fair solution to this conundrum does not require much imagination and should be in the works.
Update on Sickle Cell Disease. Since our previous discussion of sickle cell disease, the FDA has discussed (for the first time) the use of the gene-editing technique CRISPR for the treatment of sickle cell disease. The target using CRISPR is the gene BCL11A, which encodes a DNA-binding protein that regulates the substitution of fetal hemoglobin with adult hemoglobin shortly after birth. During this early developmental transition, the gamma subunit of fetal hemoglobin (alpha2-gamma2) is replaced by the beta subunit found in adult hemoglobin (alpha2-beta2). The sickle cell mutation is present in the beta subunit. Inactivation of the BCL11A gene leads to the expression of fetal hemoglobin. Currently, treatment of sickle cell patients with the drug hydroxyurea also induces fetal hemoglobin expression nonspecifically. Vertex Pharmaceuticals and CRISPR Therapeutics report that 31 of 32 patients in the current trial have had no vaso-occlusive crises nine months after treatment.
This is promising, even if fetal hemoglobin is not a perfect solution to sickle cell disease. However, the only perfect solution to sickle cell disease would be a hemopoietic stem cell transplant that replaced the mutant beta subunit with the wild-type beta subunit. Despite improvements in protocols and technique, this remains dangerous and uncertain. The drug voxelotor discussed previously is not perfect, either. But it does address the actual biochemical cause of sickle cell disease, which is the polymerization of mutant hemoglobin that leads to the sickling of red blood cells. This change in shape and stiffness causes the vaso-occlusive crises of sickle cell disease.
Obviously, it remains to be seen which intervention works best. Perhaps one treatment or the other will be found to be more effective for particular patients. The question is whether an oral drug that is well tolerated at high doses in children and adults should be preferred over an irreversible gene editing approach that has a small but finite possibility of off-target effects. These are difficult questions even in the absence of market imperatives that inevitably lead to a “can implies ought” imperative for the scientist. Or perhaps that is the other way around.
Notes
[1] Many conditions have genetic components (e.g., autoimmune diseases such as Type 1 diabetes and colon cancer), but here “genetic disease” includes only those for whom a single identifiable defective gene results in the disease. The most common autosomal dominant disease (only one mutation on one chromosome causes disease) is Familial hypercholesterolemia (LDL receptor deficiency leading to high serum cholesterol, which is one clear indication for treatment with statins). Huntington’s disease is also autosomal dominant. Common autosomal recessive diseases (mutation on both chromosomes), in rough order of incidence are sickle cell disease, cystic fibrosis, Tay-Sachs disease, and phenylketonuria. Newborn babies are screened for many of these conditions, for which early intervention can often prevent disease. Duchenne muscular dystrophy and hemophilia (HA and HB) are the two most common X-linked genetic diseases. These appear only in biological males in the absence of some other vary rare event and are sometimes referred to as sex-linked. The single mutant x chromosome in males was inherited from a mother who was a carrier, with one normal X (upper case) chromosome and one mutant x (lower case) chromosome.
[2] The two amino acid sequences are displayed here in the single letter code developed by Richard V. Eck, a remarkably versatile pioneer in the use of computers in biochemistry and molecular evolution who has been unjustly forgotten. Eck, who never finished his PhD, did not publish much but of the seven papers he published in the 1960s, five were in Science and two were in Nature. Whether Nature and Science are as good as advertised may be debatable, but the typical scientist would rejoice at one, maybe two, papers in either journal for a career. This is a record comparable to that of the late actor John Cazale, who appeared in only five full-length feature films, each of which was nominated for the Academy Award for Best Picture. The Godfather, The Godfather Part II, and The Deer Hunter won the award. The Conversation and Dog Day Afternoon did not. Each movie is a triumph of an older Hollywood.
[3] In the cell these chaperones are proteins that help a target protein find its active (native) three-dimensional structure.
[4] Triple therapy: Although the mechanisms are different here, treatments of disease with drugs that target different mechanisms responsible for the underlying pathology often work exceedingly well and at lower drug concentrations. This can also lead to fewer side effects that are likely to less harmful. The most prominent current example is HAART, highly active anti-retroviral therapy, for HIV/AIDS, in which a combination of drugs that inhibit different stages of the HIV replication cycle can essentially turn AIDS into a manageable chronic infection for most HIV-infected individuals. The only roadblock to worldwide use of HAART is financial.


How easily reproducible would this drug production be? I seem to remember that Russia has authorized Russian companies to ignore intellectual property rights if sanctions make it impossible to deal with the owner. Russia might make a lot of friends around the world by supplying this drug at a reasonable price.
This question has been addressed in Owning the Sun: A People’s History of Monopoly Medicine from Aspirin to COVID-19 Vaccines, by Alexander Zaitchik (Counterpoint Press, 2022; reviewed here at NC by yours truly). Bill Gates is reported to have led the way in protecting the “intellectual property” of Big Pharma earlier in the current pandemic. The infrastructure and technical capability to produce vaccines and drugs such as those described here is essentially worldwide…India, Brazil, Argentina, Chile, Russia, China which are largely outside the “system.” India and China already supply many, perhaps most, of our drugs. But the consequences could still be significant for the companies/countries that go rogue.